Molecular Dynamics
The collective behavior of self-assembled systems commonly gives rise to emergent properties not found within the individual building blocks. Delineating the connection between atomic-scale structure and macro-scale materials properties is not trivial, and is often difficult to precisely define using experiments alone. Complementary with our experimental studies, we employ all-atom and coarse-grained molecular dynamics (MD) simulations to further characterize our protein assemblies in silico, providing an avenue to both visualize the dynamic interplay between protein subunits at atomic resolution and investigate conditions difficult or unfeasible to achieve experimentally. Through a combination of MD, enhanced sampling (free energy) methods, and custom data analysis techniques, we seek to describe a relationship between the structure and emergent properties of self-assembled systems from the point of view of thermodynamics. The broad applicability of MD to our research makes this subgroup accessible to/from any of the others and consistently provides opportunities for creative work.
Principal members: Rob
In addition to XSEDE supercomputer resources, our lab is equipped with two home-built workstations for carrying out MD simulations:
Pauling (MD workstation)

2x Intel Xeon E5-2670 (32 cores @ 2.60 GHz)
128 GB DDR3 EEC memory
2x NVIDIA GTX 970 GPUs (3,328 CUDA cores)
512 GB SSD + 7 TB RAID1 HDD
Oppenheimer (Analysis workstation)

Intel Xeon E5-2620 (24 cores @ 2.40 GHz)
32 GB DDR4 memory
1x NVIDIA GT 730
256 GB SSD + 3 TB RAID1 HDD + 1 TB HDD scratch space
Selected Publications
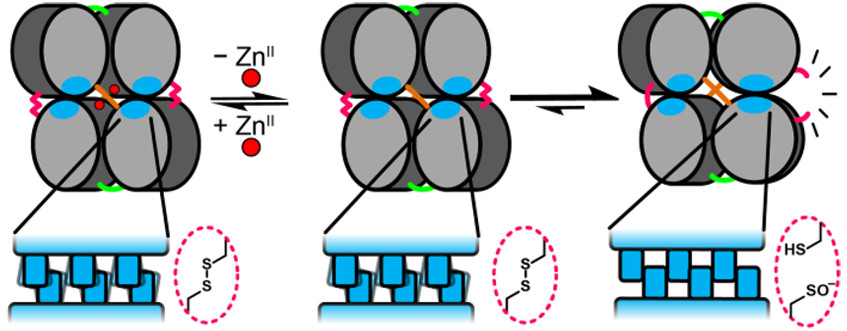
L.A. Churchfield, R.G. Alberstein, L.M. Williamson, F.A. Tezcan. Determining the Structural and Energetic Basis of Allostery in a De Novo Designed Metalloprotein Assembly, J. Am. Chem. Soc. (2018).[PDF]
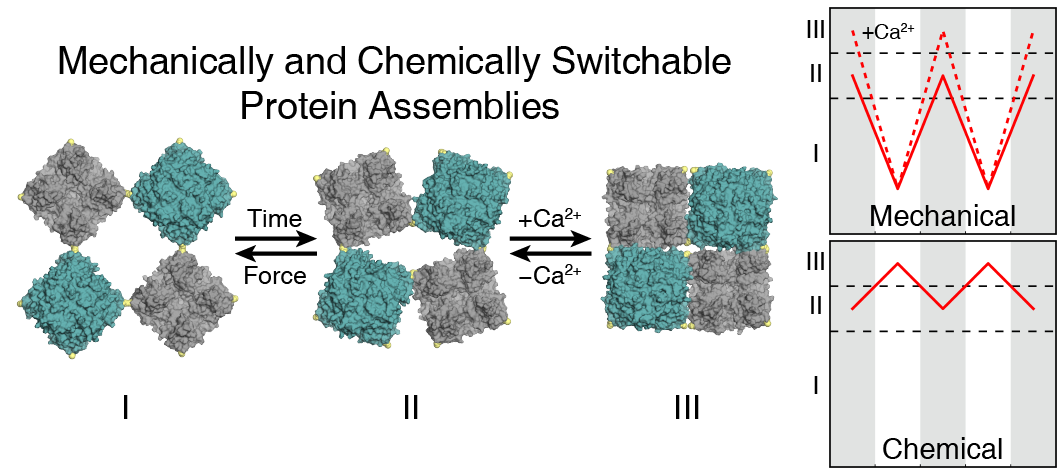
R. Alberstein, Y. Suzuki, F. Paesani, F.A. Tezcan. Engineering the entropy-driven free-energy landscape of a dynamic nanoporous protein assembly, Nat. Chem. (2018).[PDF]